




























By James R. Cheeseman and Æleen Frisch
Introduction
NMR chemical shifts are an important tool in characterizing molecular systems and structures. Accordingly, predicting NMR spectra is an essential feature of computational chemistry software. In this article, we’ll look at two very different NMR facilities, each of which is very useful when applied to the appropriate sort of molecules.
ChemDraw Ultra includes the CS ChemNMR Pro facility which can be used to estimate the 13C and 1H (proton) chemical shifts with respect to TMS. This facility is accessed from the Estimate menu within the product. When a molecule has been selected, the two items on the menu become active, and selecting one of them causes the NMR chemical shifts for the corresponding atom type to be calculated.
Figure 1 illustrates the use of this facility; here we estimate the 13C chemical shifts with respect to TMS for adenine. The Estimate menu and selected molecule appear on the left in the figure, and the resulting graphic output appears on the right. The latter consists of another copy of the molecule to which numbers denoting the chemical shift (in ppm) have been added at each atom location. Note that numbers are ordinary ChemDraw text labels and thus may be moved as needed in order to make them fully legible. Additional output is presented in text form via a Notepad document (which is opened automatically by ChemDraw).
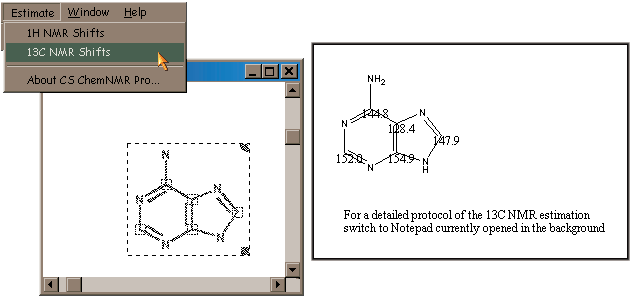
Figure 1. Example CS ChemNMR Pro Output
CS ChemNMR Pro uses a heuristically-driven procedure in order to estimate chemical shifts; it starts with a base value determined from the molecular mechanics atom type of the atom in question, and then applies corrections for each of the groups to which it is bonded in order to compute its final value. This process is illustrated in this excerpt from the textual output from a calculation on taxol:
Protocol of the 13C NMR Estimation
Node Estim. Base Incr. Comment (ppm rel. to TMS)
C 138.5 123.3 1-ethylene
-8.9 1 -C-C-C-C
-7.4 1 -C
17.3 1 -C-C-C-C
14.2 1 -C-O
The main advantage of this approach to computing chemical shifts is its speed: chemical shifts can be computed almost instantaneously even for very large molecules. However, the method has an important weakness which
must be kept in mind. Since it relies on a fixed set of parameters corresponding to atom types and subgroups, the method will be reliable only for molecules for which parameters are available and for which the assumptions about molecular structure and bonding which are built-in to the parameters are valid.
In simple terms, this NMR estimation method is appropriate only for ordinary organic molecules. It produces reasonable results for such systems, but becomes quite unreliable for systems with any unusual features: unusual bonding, strained systems, systems for which electron correlation is important for accurate modeling of the molecular structure or properties, and so on. In these cases, a more accurate computational method is required.
Last updated on: 21 June 2017.