




























Gaussian 16W Reference
A number of utility programs are included with Gaussian 16W. They are accessible via the various items on the Utilities menu in the program’s main window. We will consider them here in the same order as they appear in that menu, beginning with the NewZMat facility.
The NewZMat Facility
The NewZMat File Conversionwindow controls file conversions. It is reached via the NewZMat item on the Utilities menu. By default, it converts files to Gaussian 16W input files (.GJF files). It is an interface to the standard NewZMat utility which is included with Gaussian 16.
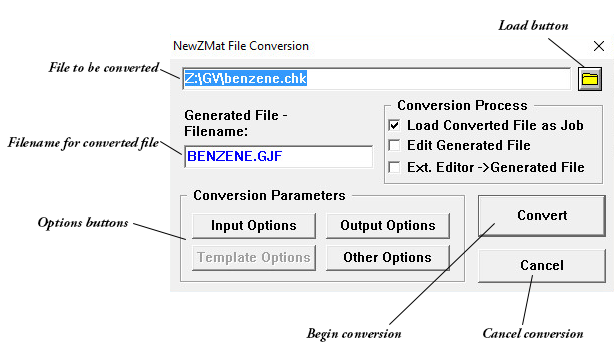
The Generated Filename field is used to specify the name of the converted file. The default is the filename of the file to be converted with the extension GJF.
The Convert button begins the conversion of the designated file, using currently set options.
The Cancel button returns you to the Job Processing window without converting any file.
The Load button (appears as a file folder icon) can be used to load a different input file to be converted.
The Conversion Process Area
Load Converted File as Job
If checked, load the converted job into memory. The default is checked if the selected file is not a .GJF file.
Edit Generated File
If checked, edit the new input file after converting using the Job Edit window (default is unchecked).
Ext. Editor → Generated File
If checked, edit the new input file after conversion using the designated external editor (default is unchecked).
Options Buttons
Input Options
Opens the NewZMat Input Options window.
Output Options
Opens the NewZMat Output Options window.
Other Options
Opens the Other Options window.
Template Options
This option is not implemented in the current version of Gaussian 16W.
The NewZMat Input Options Window
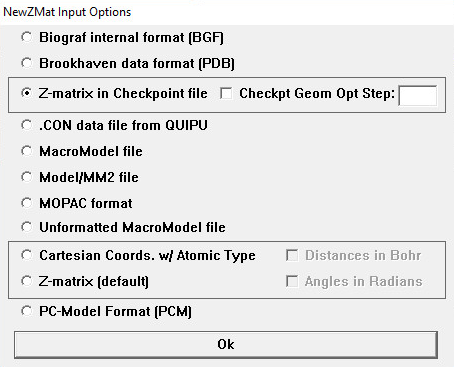
The radio buttons on this window specify the format of the file to be converted:
| Radio Button | NewZMat Option |
|---|---|
| Biograf internal file format (BGF) | -ibgf |
| Brookhaven data format (PDB) Brookhaven protein data bank format. |
-ibkv |
| Z-matrix in Checkpoint file Extract a Z-matrix from a Gaussian 16 checkpoint file, from the optimization step indicated in the Checkpt Geom Opt. Step field. |
-ichk |
| .CON data file from QUIPU | -icon |
| MacroModel file | -immodel |
| Model/MM2 | -imodel |
| MOPAC format | -imopac |
| Unformatted MacroModel file | -iummodel |
| Cartesian Coords. w/ Atomic Type | -ixyz |
| Z-matrix (this is the default) | -izmat |
| PC-Model Format (PCM) | -ipcm |
Check Boxes
Distances in Bohr
If checked (unchecked is the default), distances in the file to be converted are assumed to be in Bohr rather than angstroms (-ubohr).
Angles in Radians
If checked (unchecked is the default), angles in the file to be converted are assumed to be in radians rather than degrees (-urad).
The NewZMat Output Options Window
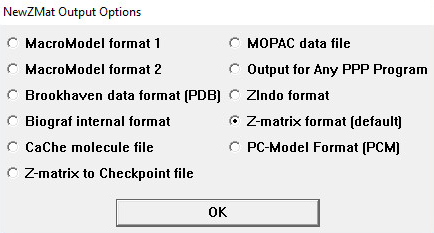
The radio buttons on this window specify the format of the generated file:
|
Radio Button
|
NewZMat Option(s) |
|---|---|
| MacroModel Format 1 | -ommodel -mof1 |
| MacroModel Format 2 | -ommodel -mof2 |
| Brookhaven data format (PDB)> | -opdb |
| Biograf internal format | -obgf |
| CaChe molecule file | -ocache |
| Z-matrix to Checkpoint file Output a Z-matrix within a checkpoint file. |
-ochk |
| MOPAC data file | -omopac |
| Output for Any PPP Program Format for a Pople-Paiser-Parr program. |
-oppp |
| ZIndo format | -ozindo |
| Z-matrix format (this is the default) | -ozmat |
| PC-model Format (PCM) | -opcm |
The NewZMat Other Options Window
This window is used to set other conversion options. The corresponding NewZMat command line options are indicated in parentheses.
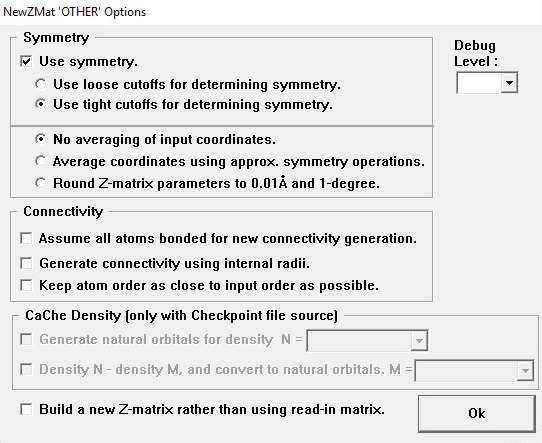
The Symmetry Area
Use symmetry
If checked (default), then use symmetry information in converting the input. (The unchecked state corresponds to -nosymm.)
Use loose cutoffs for determining symmetry
Assume molecular symmetry more easily (-lsymm).
Use tight cutoffs for determining symmetry
This is the default (-tsymm).
No averaging of input coordinates
Don’t average input coordinates (-nosymav). This is the default.
Average coordinates using approx. symmetry operations
Average input coordinates using approximate symmetry operations to achieve exact symmetry (-symav).
Round Z-matrix parameters to 0.01 Å and 1-degree
Round parameters before assigning symmetry (-round).
Connectivity Area
Assume all atoms bonded for connectivity generation
This is the default.
Generate connectivity using internal radii
Corresponds to the -gencon option.
Keep atom order as close to input order as possible
Corresponds to the -order option.
CaChe Density Area
The options in this area are available only when the input file type is checkpoint and the output file type is a CaChe molecule file.
Generate natural orbitals for density
Generate natural orbitals for the orbital specified in the N field (-density).
Density N – density M, and convert to natural orbitals
Subtract generalized density in field M from the density in field N, and then convert to natural orbitals (-mdensity).
Other Items
Build a new Z-matrix rather than use the read-in matrix
Also implies generating connectivity from internal radii (-rebuildzmat).
Debug Level
Set NewZMat internal debug level. The default is 0 (-d, one or more times).
The CubeGen Utility
This utility creates a cube file from the information stored in a Gaussian 16W checkpoint file, in a manner similar to the Cube keyword. It prompts you for the required information:
Property [Density]? MO=LUMO
Formatted Checkpoint file? water
Cube file [ ]? w_lumo.cub
Approximate points per side [0]? 75
Header in cube file [H]? N
The desired kind of cube is specified via one of the following keywords at the first prompt:
MO=n
Molecular orbital n. The keywords Homo, Lumo, All, OccA (all α occupied), OccB, Valence and Virtuals may also be used in place of a specific orbital number.
Density=type
Total density of the specified type (see below). The FDensity form requests the use of full instead of frozen core densities.
Spin=type
Spin density (α − β) of the specified type.
Alpha=type
Alpha spin density of the specified type. The FAlpha form requests the use of full instead of frozen core densities.
Beta=type
Beta spin density of the specified type. The FBeta form requests the use of full instead of frozen core densities.
Potential=type
Electrostatic potential of the specified type.
type is one of the single density selection options that are valid with the Gaussian 16W Density keyword: HF, MP2, CI, QCI, and so on (Current is not supported).
The Approximate points per side prompt specifies the grid size: a rectangular grid of n3 evenly distributed points, which may or may not be a cube. The default is 80 if 0 is specified for this prompt. If the value of -1 is given in response to the prompt, then the grid specifications will be accepted after the final prompt, as with Cube=Cards.
The keywords Coarse, Medium and Fine may also be used to specify values of 40, 80 and 100, respectively.
The CubMan Utility
The cubman program manipulates cubes of values of electron density and electrostatic potential as produced by Gaussian. The program prompts for an operation to perform, and then the names of the necessary files. The possible operations and their associated subcommands are:
- add: Add two cubes to produce a new one.
- copy: Copy a cube, possibly converting it from formatted to unformatted or vice versa.
- diff: Compute properties of the difference between two cubes, without writing out a new cube.
- prop: Computes the properties of a single cube.
- subtract: Subtracts two cubes to produce a new cube.
- scale: Scale a cube by a constant factor, producing a new cube.
All operation subcommands can be abbreviated to the shortest unique form.
Here are two annotated sample runs with cubman (user input is shown in boldface type, and output has been condensed
slightly due to space considerations):
Action [Add, Copy, Difference, Props, SUbtract, SCale]? p Input file? b.cube Is it formatted [no,yes,old]? y Gaussian cube files are formatted Opened special file b.cube. Input file titles: First excited state of propellane Title line from the job CI Total Density Contents of cube file SumAP= 13.39263 SumAN= .00000 SumA= 13.39263 Stats about cube CAMax= 3.35320 XYZ= .18898 -1.32280 .000004 CAMin= .00000 XYZ= -9999.00000 -9999.00000 -9999.00000 DipAE= -.8245357658 .7624198057 .1127178115 DipAN= -.0000060000 -.0000060000 .0000000000 DipA= -.8245417658 .7624138057 .1127178115 Action [Add, Copy, Difference, Props, SUbtract, SCale]? su First input? b.cube Is it formatted [no,yes,old]? y Opened special file b.cube. Second input? a.cube Is it formatted [no,yes,old]? y Opened special file a.cube. Output file? c.cube File to hold the new cube Should it be formatted [no,yes,old]? y Opened special file c.cube. Input file titles: First excited state of propellane Title from first file CI Total Density Contents of first cube Input file titles: Propellane HF/6-31G* Title from second file SCF Total Density Contents of second cube Output file titles: Title used for new file First excited state of propellane || Propellane HF/6-31G* CI Total Density - SCF Total Density Difference to be computed SumAP= 13.39263 SumAN= .00000 SumA= 13.39263 Stats for 1st cube CAMax= 3.35320 XYZ= .18898 -1.32280 .000004 CAMin= .00000 XYZ= -9999.00000 -9999.00000 -9999.00000 SumBP= 13.38168 SumBN= .00000 SumB= 13.38168 Stats for 2nd cube CBMax= 3.39683 CBMin= .00000 SumOP= .63453 SumON=-.62358 SumO= .01094 Stats for new cube COMax= .49089 COMin=-.39885 DipAE= -.8245357658 .7624198057 .1127178115 DipAN= -.0000060000 -.0000060000 .0000000000 DipA= -.8245417658 .7624138057 .1127178115 DipBE= -.8306292172 .5490287046 .1243830393 DipBN= -.0000060000 -.0000060000 .0000000000 DipB= -.8306352172 .5490227046 .1243830393 DipOE= .0060934514 .2133911011 -.0116652278 DipON= -.0000060000 -.0000060000 .0000000000 DipO= .0060874514 .2133851011 -.0116652278
In the output. the input cubes are denoted as A and B, and the output cube is designated by O. Other code letters are N for “negative values” or for “nuclear,” depending on the context, P for “positive values,” E for “electronic,” C for “charge,” Dip for “dipole,” Sum for “sum,” Max for “maximum,” and Min for “minimum.” Thus, SumAN is the sum over the first input cube, taking the negative values only, and DipON is the nuclear contribution to the dipole moment for the output cube. Similarly, CBMax is the maximum charge for the second input cube, and SumO is the sum of the values in the output cube, including both positive and negative values.
The FreqChk Utility
FreqChk is used to generate frequency and thermochemistry data from a checkpoint file. Here is an example of its use in this mode:
Checkpoint file? water Write Hyperchem files? N Temperature (K)? [0=>298.15] 300 Specify desired temperature. Pressure (Atm)? [0=>1 atm] 0 Specified desired pressure. Scale factor for frequencies during thermochemistry? [0=>1/1.12] 0.9613 Specify scale factor for thermochemical calculation. Do you want to use the principal isotope masses? [Y]: N Use this feature to substitute other isotopes for the standard (most abundant) ones. For each atom, give the integer mass number. In each case, the default is the principal isotope. Atom number 1, atomic number 8: [16] 16 Atom number 2, atomic number 1: [1] 2 Atom number 3, atomic number 1: [1] 2------------------------------------------------------------ Center Atomic Coordinates (Angstroms) Number Number X Y Z------------------------------------------------------------ 1 8 0.000000 0.000000 0.058070 2 1 0.000000 0.407058 -0.232281 3 1 0.000000 -0.407058 -0.232281------------------------------------------------------------ Atom 1 has atomic number 8 and mass 15.99491 Atom 2 has atomic number 1 and mass 2.01410 Atom 3 has atomic number 1 and mass 2.01410 … 1 2 A1 A1 Frequencies — 2170.0510 4142.4280 Red. masses — 1.07851.0491 Frc consts — 2.9923 10.6067 IR Inten — 7.2428 44.3099 Raman Activ — 9.2625 47.7624 Depolar — .7245 .1791 Atom AN X Y Z X Y Z 1 8 .00 .00 .07 .00 .00 .05 2 1 .00 -.45 -.54 .00 .57 -.42 3 1 .00 .45 -.54 .00 -.57 -.42 ------------------- - Thermochemistry - ------------------- Temperature 300.000 Kelvin. Pressure 1.00000 Atm. Thermochemistry will use frequencies scaled by 0.9613. Molecular mass: 20.02312 amu. Zero-point correction= 0.060789 Thermal correction to Energy= 0.063639 Thermal correction to Enthalpy= 0.064589 Thermal correction to Gibbs Free Energy= 0.043825 …
Note that when a scale factor is included, the frequencies are scaled only when they are used to perform the thermochemistry analysis. The displayed frequencies themselves are not scaled.
The FormChk Utility
This utility produces an ASCII formatted checkpoint file from a Gaussian 16W checkpoint file. Formatted checkpoint files are the recommended method for transferring data to graphics and other post-processing programs.
Here is an example use of FormChk:
Checkpoint file? water
Read checkpoint file water.chk
Write formatted file water.fch
Note that formatted checkpoint files have the extension .FCH in the Windows environment.
The UnFchk Utility
This utility is the opposite number to FormChk. It converts a formatted checkpoint file to a binary Gaussian 16W checkpoint file:
Formatted Checkpoint file? water
Read formatted file water.fch
Write checkpoint file water.chk
The ChkChk Utility
This utility displays the route and title sections corresponding to a checkpoint file and indicates other information that is present within it:
Checkpoint file? water
Checkpoint file water.chk:
Title: Water frequencies
Route: #T B3LYP/6-31G* Freq Test
Atomic coordinates present.
Z-matrix present with variables.
MO coefficients present.
Cartesian force constants present.
Internal force constants may be present.
The C8616 Utility
This utility converts binary checkpoint files between the formats used by Gaussian 16Wand previous program versions. Note that the output from C8616is not intended to be readable. Use the FormChk utility to produce human- and program-readable formatted checkpoint files.
Last updated on: 11 May 2017.