




























In commemoration of its 120th anniversary, The Journal of Physical Chemistry recently announced a list of its top 25 cited articles. Papers by several of Gaussian’s developers (whose names are in bold) appear in the collection:
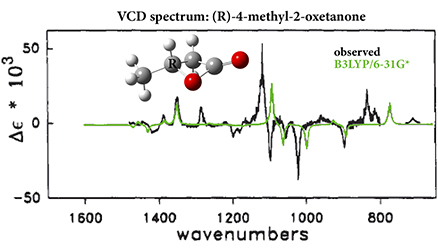
#2: “Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields,” P. J. Stephens, F. J. Devlin, C. F. Chabalowski, M. J. Frisch, J. Phys. Chem., 1994, 98, 11623-11627. DOI: 10.1021/j100096a001
Uses the just-introduced B3LYP functional to predict the unpolarized absorption and circular dichroism spectra of the fundamental vibrational transitions (VCD) of 4-methyl-2-oxetanone.
#10: “Reaction path following in mass-weighted internal coordinates,” Carlos Gonzalez, H. Bernhard Schlegel, J. Phys. Chem., 1990, 94, 5523-5527. DOI: 10.1021/j100377a021
Presents a new method for computing reaction paths (IRCs) in mass-weighted internal coordinates.
#22: “Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions,” Aleksandr V. Marenich, Christopher J. Cramer, and Donald G. Truhlar, J. Phys. Chem., 2009, 113, 6378-6396. DOI: 10.1021/jp810292n
Features the SMD model, a new continuum solvation model that employs the quantum mechanical charge density of a solute molecule reacting to a continuum description of a solvent as its basis.
#25: “Toward a systematic molecular orbital theory for excited states,” James B. Foresman, Martin Head-Gordon, John A. Pople, Michael J. Frisch, J. Phys. Chem., 1992, 96, 135-149. DOI: 10.1021/j100180a030
Describes CI-Singles, a new method for predicting the ab initio energy, wavefunction, and the gradient of a molecule in an electronically excited state.
We congratulate these researchers on their accomplishments.