




























- Model Chemistries
- Job Types
- Program Limits
- Links
Model Chemistries
The combination of method and basis set specifies a model chemistry to Gaussian, specifying the level of theory. Every Gaussian job must specify both a method and basis set. This is usually accomplished via two separate keywords within the route section of the input file, although a few method keywords imply a choice of basis set. Some jobs using a density functional method may also include a density fitting set (see the Basis Sets for more information).
The following table lists methods which are available in Gaussian, along with the job types for which each one may be used. An asterisk indicates analytic calculations, while numerical-only calculations are indicated by num (see the discussion of the specific keyword in question for details).
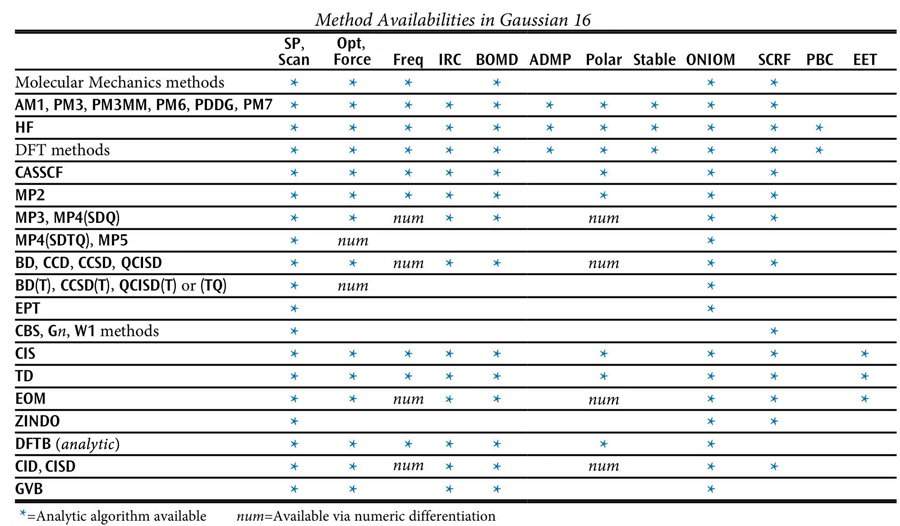
If no method keyword is specified, HF is assumed. Most method keywords may be prefaced by R for closed-shell restricted wavefunctions, U for unrestricted open-shell wavefunctions, or RO for restricted open-shell wavefunctions: for example, ROHF, UMP2, or RQCISD. RO is available only for Hartree-Fock and Density Functional methods, and AM1, PM3, PM3MM, PM6, and PDDG energies and gradients, and MP2, MP3, MP4, and CCSD energies.
In general, only a single method keyword should be specified, and including more than one of them will produce bizarre results. However, there are exceptions:
- CASSCF may be specified along with MP2 to request a CASSCF calculation including dynamic electron correlation.
- ONIOM and IRCMax jobs require multiple method specifications. However, they are given as options to the corresponding keyword.
- The form model2 // model1 described in Job Type may be used to generate an automatic optimization followed by a single point calculation at the optimized geometry.
Job Types
The following table lists the job types available in Gaussian 16:
- SP: Single point energy.
- Opt: Geometry optimization.
- Freq: Frequency and thermochemical analysis.
- IRC: Reaction path following.
- IRCMax: Find the maximum energy along a specific reaction path.
- Scan: Potential energy surface scan.
- Polar: Polarizabilities and hyperpolarizabilities.
- ADMP and BOMD: Direct dynamics trajectory calculation.
- EET: Excitation energy transfer calculation.
- Force: Compute forces on the nuclei.
- Stable: Test wavefunction stability.
- Volume: Compute molecular volume.
- Density=Checkpoint Guess=Only: Recompute population analysis only.
- Guess=Only: Print initial guess only; generate fragment-based initial guess.
In general, only one job type keyword should be specified. The exceptions to this rule are:
- Polar and Opt may be combined with Freq. In the latter case, the geometry optimization is automatically followed by a frequency calculation at the optimized structure.
- Opt may be combined with the compound method keywords in order to specify options for the optimization portion of the calculation: e.g., Opt=(TS,ReadFC) CBS-QB3.
When no job type keyword is specified within the route section, the default calculation type is usually a single point energy calculation (SP). However, a route section of the form: method2/basis2 // method1/basis1 may be used to request an optimization calculation (at method1/basis1) followed by a single point energy calculation (at method2/basis2) at the optimized geometry. For example, the following route section requests a B3LYP/6-31G(d) geometry optimization followed by a single point energy calculation using the CCSD/6-31G(d) model chemistry:
# CCSD/6-31G(d)//B3LYP/6-31G(d)
In this case, the Opt keyword is optional and is the default. Note that Opt Freq calculations may not use this syntax.
Molecular Properties
The following table provides a mapping between commonly-desired predicted quantities and the Gaussian 16 keywords that will produce them:
- Anharmonic IR/Raman/VCD/ROA spectra: Freq=Anharmonic
- Antiferromagnetic coupling: Guess=Fragment, Stable
- Atomic charges: Pop
- ΔG of solvation: SCRF=SMD
- Dipole moment: Pop
- Electron affinities: CBS-QB3, CCSD, EPT
- Electron density: cubegen
- Electronic circular dichroism: CIS, TD, EOM, SAC-CI
- Electrostatic potential: cubegen, Prop
- Electrostatic potential-derived charges: Pop=Chelp, ChelpG or MK
- Electronic transition band shape: Freq=FranckCondon, Freq=HerzbergTeller
- Polarizabilities/hyperpolarizabilities: Freq, Polar, CPHF=RdFreq, Polar=DCSHG
- High accuracy energies: CBS-QB3, G2, G3, G4, W1U, W1BD
- Hyperfine coupling constants (anisotropic): Prop
- Hyperfine spectra tensors (including g tensors): Freq=(VCD, VibRot, Anharmonic)
- Ionization potentials: CBS-QB3, CCSD, EPT
- IR and Raman spectra: Freq
- Pre-resonance Raman spectra: Freq CPHF=RdFreq
- Resonance Raman spectra: Freq=ReadFCHT
- Molecular orbitals: Pop=Regular
- Multipole moments: Pop
- NMR shielding and chemical shifts: NMR
- NMR spin-spin coupling constants: NMR=Mixed
- Optical rotations: Polar=OptRot
- Raman optical activity: Freq=ROA
- Thermochemical analysis: Freq
- UV/Visible spectra: CIS, ZIndo, TD, EOM, SAC-CI
- Vibration-rotation coupling: Freq=VibRot
- Vibrational circular dichroism: Freq=VCD
- Vibronic spectra: Freq=ReadFCHT
Program Limitations
This section lists the various size limitations that exist within Gaussian 16.
- The integral program has the following limitations:
- The maximum number of atoms is 250,000.
- The maximum total number of primitive shells is 750,000.
- The maximum number of primitive d-shells and higher is 250,000.
- The maximum number of contracted shells is 250,000.
- The maximum degree-of-contraction allowed is 100.
- Opt=(EF,EnOnly) optimizations—useful only for methods without analytic gradients—are limited to 50 variables.
- The GVB program is limited to 100 paired orbitals (which is not a restriction in practice).
- The internal version of NBO 3 is dimensioned for 250,000 atoms and 10,000 basis functions.
Links
The following are the component programs of Gaussian 16—known as links—along with their primary functions:
- L0: Initializes program and controls overlaying
- L1: Processes route section, builds list of links to execute, and initializes scratch files
- L101: Reads title and molecule specification
- L102: Fletcher-Powell optimizations
- L103: Berny optimizations to minima and TS, STQN transition state searches
- L105: Murtaugh-Sargent optimizations
- L106: Numerical differentiation of forces/dipoles to obtain polarizability/ hyperpolarizability
- L107: Linear-synchronous-transit (LST) transition state search
- L108: Unrelaxed potential energy surface scan
- L109: Newton-Raphson optimization
- L110: Double numerical differentiation of energies to produce frequencies
- L111: Double numerical differentiation of energies to compute polarizabilities and hyperpolarizabilities
- L112: Performs the Self-Consistent Virial Scaling method (SCVS), T. A. Keith's extension of [Lowdin59, Magnoli82, Lehd91]
- L113: EF optimization using analytic gradients
- L114: EF numerical optimization (using only energies)
- L115: Follows reaction path using GS3 algorithm
- L116: Numerical self-consistent reaction field (SCRF)
- L117: Performs IPCM solvation calculations.
- L118: BOMD calculations
- L120: Controls ONIOM calculations
- L121: ADMP calculations
- L122: Counterpoise calculations
- L123: Follows reaction path using the HPC algorithm (and others)
- L124: Performs ONIOM with PCM and external-iteration PCM
- L202: Reorients coordinates, calculates symmetry, and checks variables
- L301: Generates basis set information
- L302: Calculates overlap, kinetic, and potential integrals
- L303: Calculates multipole integrals
- L308: Computes dipole velocity and Rx∇ integrals
- L310: Computes spdf 2-electron integrals in a primitive fashion
- L311: Computes sp 2-electron integrals
- L314: Computes spdf 2-electron integrals
- L316: Prints 2-electron integrals
- L319: Computes 1-electron integrals for approximate spin orbital coupling
- L401: Forms the initial MO guess
- L402: Performs semi-empirical and molecular mechanics calculations
- L405: Initializes an MCSCF calculation
- L502: Iteratively solves the SCF equations (conven. UHF & ROHF, all direct methods, SCRF)
- L503: Iteratively solves the SCF equations using direct minimization
- L506: Performs an ROHF or GVB-PP calculation
- L508: Quadratically convergent SCF program
- L510: MC-SCF
- L601: Population and related analyses (including multipole moments)
- L602: 1-electron properties (potential, field, and field gradient)
- L604: Evaluates MOs or density over a grid of points
- L607: Performs NBO analyses
- L608: Non-iterative DFT energies
- L609: Atoms in Molecules properties
- L610: Numerical integration (for testing integral codes)
- L701: 1-electron integral first or second derivatives
- L702: 2-electron integral first or second derivatives (sp)
- L703: 2-electron integral first or second derivatives (spdf)
- L716: Processes information for optimizations and frequencies
- L801: Initializes transformation of 2-electron integrals
- L802: Performs integral transformation (N3 in-core)
- L804: Integral transformation
- L811: Transforms integral derivatives & computes their contributions to MP2 2nd derivatives
- L901: Anti-symmetrizes 2-electron integrals
- L902: Determines the stability of the Hartree-Fock wavefunction
- L903: Old in-core MP2
- L904: Complete basis set (CBS) extrapolation method of Petersson, et. al.
- L905: Complex MP2
- L906: Semi-direct MP2
- L908: Electron Propagator Program
- L909: ADC(3) and related electron propagator models
- L913: Calculates post-SCF energies and gradient terms
- L914: CI-Singles, RPA and ZIndo excited states; SCF stability
- L915: Computes fifth order quantities (for MP5, QCISD(TQ) and BD(TQ))
- L916: Old MP4 and CCSD
- L918: Reoptimizes the wavefunction
- L923: SAC-CI program
- L925: Implements the Excited State Electron Transfer (EET) model
- L1002: Iteratively solves the CPHF equations; computes various properties (including NMR)
- L1003: Iteratively solves the CP-MCSCF equations
- L1014: Computes analytic CI-Singles second derivatives
- L1101: Computes 1-electron integral derivatives
- L1102: Computes dipole derivative integrals
- L1110: 2-electron integral derivative contribution to F(x)
- L1111: 2 particle density matrix and post-SCF derivatives
- L1112: MP2 second derivatives
- L9999: Finalizes calculation and output
Last updated on: 02 May 2017. [G16 Rev. C.01]