by Æleen Frisch and Michael J. Frisch
FOX-7 is an explosive compound first synthesized in 1998 [Ostmark98, Bemm98, Latypov98] by the Swedish National Defense Research Institute, now the Swedish Defense Research Agency (FOI). This compound is a result of the organization’s ongoing research effort to develop explosives that are “more powerful, safer and environmentally friendlier” [Ostmark06] than existing alternatives. The goal of such research is to synthesize compounds that are powerful explosives yet are insensitive to heat, fire, shock and impact and other predictable hazards of armed conflict environments. FOX-7 is an effective explosive with detonation velocity of ~8335 m/s [Karlsson02] (cf. TNT at 6900 m/s, octanitrocubane at 10100 m/s), combining high performance with low sensitivity.
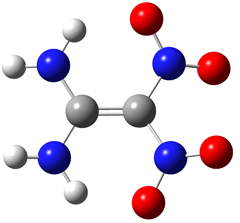
The FOX-7 molecule is pictured above: C2(NH2)2(NO2)2. Its IUPAC name is 2,2-dinitroethene-1,1-diamine (DADNE). As reported in [Bemm98], the carbon and amino nitrogen atoms are essentially planar while the nitro nitrogen atoms are out of plane. The molecule includes two intramolecular hydrogen bonds between the amino and nitro groups. In the solid phase, the packing pattern consists of wave-shaped layers: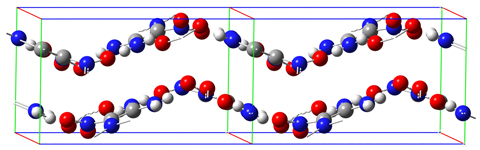
FOX-7 dimer has received attention recently due to its inclusion in the Army Research Laboratory-University of Delaware (ARL-UD) blind benchmark set [Taylor16] for testing DFT models’ accuracy in predicting intermolecular interaction energies. It is a molecule that is well suited to such an evaluation and proves challenging for many model chemistries.
Ju and coworkers published a theoretical study of the gas phase dimer and the crystal [Ju03]. Using the B3LYP functional and a series of basis sets culminating with 6-311++G(d,p), they located four stable minima for FOX-7 dimer, denoted I through IV:
- Form I has Ci symmetry; the two monomers are stacked parallel to one another, with significant overlap between their projections. There are two hydrogen bonds across the monomers: amino oxygen atoms to nitro hydrogen atoms.
- Form II is a variation of I.
- Form III has C2 symmetry. The complex has hydrogen bonds involving the amino oxygen atoms in both monomers.
- Form IV has the same wave-shape as the packing form in the crystal and C1 symmetry. There are three intermolecular hydrogen bonds across the monomers.
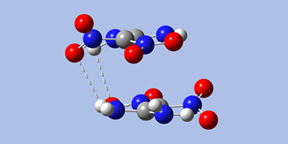 |
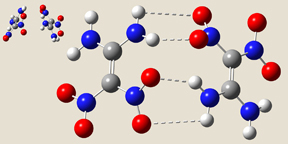 |
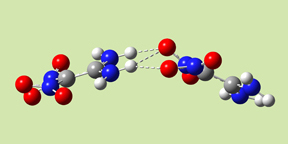 |
| Form I | Form III | Form IV |
In this study, IV had the lowest energy, with the other forms very close in energy, differing only by 1.83 kcal/mol for form III and by ~3.1 kcal/mol for forms I and II (zero-point and counterpoise corrected energies, with the ZPE scaled by 0.96). The structure included in the ARL-UD test set is very close to IV; the two longer hydrogen bond lengths in the former are somewhat shorter than in the latter: 2.19Å and 2.22Å vs. 2.30Å and 2.33Å.
The B3LYP functional does not include treatment of dispersion, which is often an important effect for systems like FOX-7 dimer. We started from structures having the general form of I, III and IV and optimized them using the APFD functional which includes dispersion and with the MP2 method, using the 6-311+G(2d,p) and aug-cc-pVDZ basis sets, respectively. We also performed B3LYP/6-311+G(2d,p) optimizations on the same structures. In some cases, we optimized the ARL-UD structure in addition to IV (see table below). All optimizations were unconstrained, and they were followed by frequency calculations to characterize the stationary points. Thermal corrected energies are used for all comparisons. All calculations were performed in Gaussian 16 Rev A.03.
The results of our calculations are summarized in the table below. They indicate that a proper treatment of weak interactions is important when modeling this molecular system. Our B3LYP/6-311+G(2d,p) calculations resulted in only two distinct minima. The I and III form starting structures both converged to a structure similar to III. The ARL-UD starting structure converged to a minimum 2.33 kcal/mol lower in energy having form IV.
The results of the optimizations with the APFD functional and the MP2 method are in agreement and quite different from the B3LYP results. In fact, a conformation like form IV cannot be located. In fact, a frequency calculation of the ARL-UD form (characterized as an MP2/aug-cc-pVDZ optimized structure) reveals this stationary point to be a transition structure and not a minimum. Distorting it in the direction of the imaginary mode and reoptimizing leads to a minimum having form I. The optimization starting from form IV also locates a structure of form I which is 1.09 kcal/mol lower in energy.
The APFD results are similar to MP2. Three minima with structures like form I were located, all lying <1 kcal/mol from one another. The lowest energy APFD structure is similar to the MP2 minimum, although the hydrogen bond lengths vary. In the APFD structure, both hydrogen bonds are 2.75Å; in the MP2 structure, they are 2.59Å and 2.63Å.
| Starting form | B3LYP/6-311+G(2d,p) | APFD/6-311+G(2d,p) | MP2/aug-cc-pVDZ | |||
|---|---|---|---|---|---|---|
| optimized form | ΔEtherm | optimized form | ΔEtherm | optimized form | ΔEtherm | |
| I | III | 2.33 | ||||
| III | III | 2.33 | I | 0.0 | ||
| IV | IV | 0.0 | I | 0.88 | I | 0.0 |
| ARL-UD | I | 0.68 | I | 1.09 | ||
The minimum found by APFD and MP2, with its parallel planes with a large amount of overlap of the two monomers, can also be related to the crystal structure, taking monomers from adjacent layers rather than adjacent molecules within a single layer. Of course, the question as to whether FOX-7 dimer exists in the gas phase is an open one.

Last updated on: 14 February 2018.